План
- Технология производства соляной кислоты
- Производство азотной кислоты
- Производство серной кислоты
- Производство полимеров
- Производство химических волокон
- Производство пластмасс
- Синтезы на основе ацетилена
- Подготовка угля к коксованию
- Получение синтезированного газа
- Список использованной литературы
1. Технология производства соляной кислоты
Соляная кислота—бесцветная жидкость, представляющая собой раствор хлористого водорода в воде. Она энергично растворяет многие металлы и их окислы. В технике применяется как соляная кислота, так и хлористый водород.
Хлористый водород используют для производства хлорорганических продуктов путем гидрохлорировании органических соединений, например этилена , ацетилена.
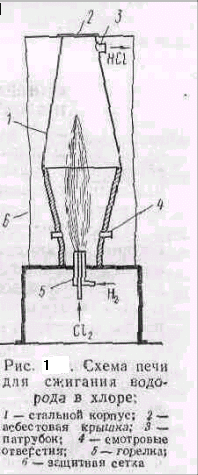 (Соляную кислоту применяют для получения хлоридов Zn, Ba. Mg, Са, Fe, A1 и т. д., для травления при пайке и лужении, и цветной металлургии (извлечение Pt, An), при гидролизе древесины, в производетве красителей, для гидрохлорировании органических соединении и т. д.
(Соляную кислоту применяют для получения хлоридов Zn, Ba. Mg, Са, Fe, A1 и т. д., для травления при пайке и лужении, и цветной металлургии (извлечение Pt, An), при гидролизе древесины, в производетве красителей, для гидрохлорировании органических соединении и т. д.
Процесс получения соляной кислоты имеет две стадии;
1) получение хлористого водорода;
2) абсорбция xлористого водорода водой.
Существуют два способа получения хлористого водорода: сульфатной и синтетический. Кроме того, в производстве; соляной кислоты используют хлористый водород, являющийся отходом при хлорировании насыщенных углеводородов
Сульфатный способ получения хлористого водорода основан на взаимодействии твердой поваренной соли с серной кислотой
2NaCl + H2SO4 = Na2SO4 + 2НС1 — Q.
Процесс синтеза проводят в муфельных печах при температуре 500—550° С, обогреваемых через стенку топочными газами. Концентрация хлористого водорода н газе от 30 до 50% НС1. Способ находит применение,но новых производств не организуют.
При синтетическом способе синтез хлористого водорода проводится по реакции
Н2 + С12—” 2НС1 + 184,33 кдж
Cyxoй хлор и водород при нормальных условиях и в темноте не реагируют между собой, но на свету или при нагревании в присутствии паров воды реакция взаимодействия их сопровождается взрывом.
Синтез проводят при избытке 5—10% водорода и высокой температуре в печи, корпус которой изготовляют из углеродистой или легированной стали, а крышку из асбеста. Нижняя часть печи (рис.1) выложена огнеупорным материалом. В ней помещена горелка, состоящая из двух концентрически расположенных стальных трубок. По наружной трубке в печь подается водород, а по внутренней — хлор. Которые на выходе из горелки смешиваются и спокойно реагируют; образуя факел горения с температурой 2000—2400° С.
Абсорбция хлористого водорода в воде идет с выделением большого количества тепла (образование гидратов), которого достаточно для нагревания кислоты до кипения. Для получения более концентрированной соляной кислоты необходим отвод тепла, так как растворимость хлористого водорода в воде с повышением температуры уменьшается.
Поглощение НС1 проводят в абсорберах с отводом тепла. Через стенку (изотермическая абсорбция) или с отводом тепла в результате испарения части воды (адиабатическая абсорбция).
Соляная кислота выпускается следующих сортов: техническая (27,5% НС1); синтетическая (31% НС1), ингибиторная (20% НС1) и реактивная (35—38% НС1, плотность при 20°С равняется 1,17— 1,19 г/см3).
Кислоту перевозят и хранят в гуммироваявых стальных цистернах и контейнерах, в фаолитовых баках и контейнерах и в емкостях, изготовленных из керамики и стекла При введении в кислоту 1—3% ингибитора активность НС1 к стали снижается в 150—200 раз, поэтому ингибиторную кислоту перевозят в стальных нефутерованных цистернах.
2.Производство азотной кислоты
Безводная азотная кислота HNO3—тяжелая бесцветная жидкость плотностью 1520 кг/м3(при 15° С). Она замерзает при температуре —47° С и кипит при 85°С, При кипении HNO3 частично разлагается с выделением двуокиси азота. С водой HNO3 смешивается в любых соотношениях, выделяя тепло, а с двуокисью азота образует нитроолеум.
 Концентрированная кислота не реагирует с алюминием, хромом и даже железом, поэтому аппаратуру для получения азотной кислоты готовят из xpoмоникелевых сталей, алюминия или из стали, футерованной кислотоупорной керамикой.
Концентрированная кислота не реагирует с алюминием, хромом и даже железом, поэтому аппаратуру для получения азотной кислоты готовят из xpoмоникелевых сталей, алюминия или из стали, футерованной кислотоупорной керамикой.
Получение слабой азотной кислоты имеет две стадии;
а) окисление аммиака до окиси азота N0;
б) переработка N0 в азотную кислоту.
Окисление аммиака проводятся при температуре 800—900°С в присутствии катализатора, изготовленного из сплава платины н родия (5—10%) в виде сеток, сплетённых из тонкой проволоки. Кроме платины, могут применяться менее активные катализаторы на основе окиси кобальта или железа с активирующими добавками. Аммиак может окисляться при 900°С и безкатализатора, но в этом случае получаетсяне окись азота, а азот:
4NH3 + 3О2=2N3 + 6Н20 + Q.
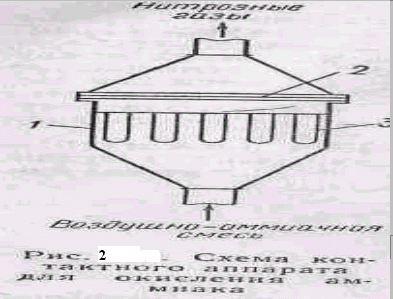
Катализаторы очень чувствительны к примесям сероводорода, пыли и т, д., поэтому воздух перед смешением с аммиаком тщательно очищается. На рис. 2 показана схема контактного аппарата для окисления аммиака под атмосферным давлением. Аппарат имеет корпус цилиндрической формы в ней закреплены платино-радиовые сотки (3—4 шт.) и поролитовые трубки (из пористой керамики) для очистки воздушко-амиачной смеси от пыли. Для получения окиси азота в контактный аппарат подают смесь, содержащую 10—11%. Повышать содержание амиака нельзя, так как при 20° С смесь с содержанием 15—28% NH3 становится взрывоопасной. При прохождении cmcеcи через платиновые сетки аммиак окисляется с образованием N0. Степень окисления аммиака составляет 98%.
Контактные аппараты, работающие под давлением 1,5—10 am (9,81•104н/м2), мало отличаются от описанных выше, но в них имеется 16—20 сеток и аппаратура более толстостенная.
Переработка окиси азота в разбавленную азотную кислоту осуществляется следующим образом. Выходящие из контактного аппарата нитрозные газы охлаждаются, и окись азота N0 окисляется самопроизвольно кислородом:
2NO +О2 - > 2NO2 + Q.
Окисление NO в NO2 происходит очень медленно. Для увеличения скорости окисления необходимо понижать температуру (реакция аномальная, скорость растет при снижении температуры в отличие от других реакции) и испытать дарение (при увеличении давления с 1 до 10 am скорость возрастает в 100U раз). Поэтому окисление N0 в N02 и абсорбцию NO2 часто происходит в установках, работающих под давлением 1,5—10 am (0,15—1 Мн/м2), что резко сокращает объемы окислительно-абсорбционных башен.
Абсорбция двуокиси азота осуществляетсяводой по суммарному уравнению
3NO2+ Н2О - > 2НNО3 + NO + Q.
 Нитрит натрия затем окисляется до нитрата натрия.
Нитрит натрия затем окисляется до нитрата натрия.
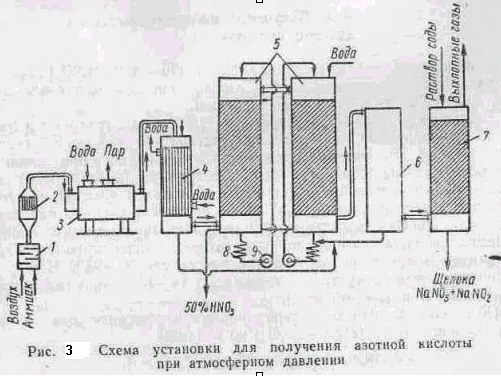
На рис. 3 показана принципиальная схема получения азотной кислоты при атмосферном давлении. Воздух и аммиак после очистки от примесей подаются в смеситель1, а затем в контактный аппарат 2. Для окисления амиака. Образовавшиеся нитрозные газы при температуре 800°С выходят из аппарата и, пройдя котел-утилизатор 3, oхлаждаются до 250°С и поступают в кожухотрубный холодильник4, где дополнительно охлаждаются до 30° С, В холодильнике начитаются окисление N0 до NO2 и конденсация пapoв, воды, при этом частично образуется HNO3
Из холодильника нитрозные газы направляются в абсорбционные насадочные башни 5, в которых окислы азота поглощаются водой; таких башен в системе б—8 шт. Прейдя последовательно через эти башни, газы поступают в окислительную башню б, где оставшаяся часть N0 окисляется в NO2 и затем а башни щелочной абсорбции 7. Для поглощения N0 последняя башня орошается водой. Образовавшаяся слабая кислота охлаждается в холодильниках 8 и с помощью насосов 9 проходит последовательно противотоком газу все поглотительные башни. Кислота (50% HNO3) выводится из первой по ходу газа башни. Степень переработки окислов азота в азотную кислоту составляет 92%, а остальные окислы азота улавливаются в башнях щелочной абсорбции.
В установках, работающих под давлением 1,5—10 am (0,15— 1 Мн/м2} и по комбинированной схеме, степень поглощения окислов азота водой составляет 99%, а получаемая кислота более крепкая — 60—62%.
3. Производство серной кислоты
Моногидрат— серная кислота (100% H2SO4) представляет собой бесцветную маслянистую жидкость плотностью 1830,3 кг/м3, кипящую при 296,2s С и атмосферном давлении и замерзающуюпри+10,45° С.
В технике серной кислотой называют не только моногидрат, но и растворы его в воде различной концентрации H2SO4 +Н20. Раствор серного ангидрида SO4 в моногидрате называют олеумом H2SO4 + SO3.При применении, транспортировке и производстве необходимо знать температуры плавления и кипения серной кислоты. При повышении концентрации серной кислоты от 0 до 64,35° до 100% образуются шесть индивидуальных химических соединений (гидратов), которые в твердом виде взаимно нерастворимы, а образуют эвтектические смеси.
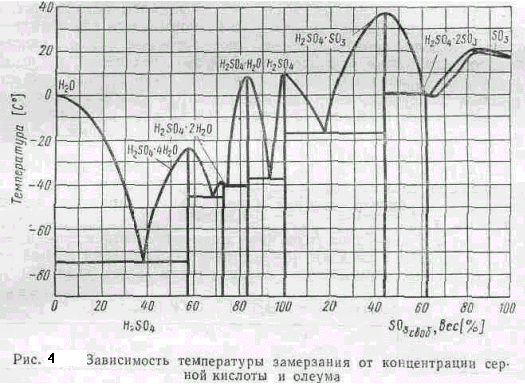 С увеличением концентрацииSO3 от 64,35% до 100% при кристаллизации образуются твердые растворы. Все сорта выпускаемой кислоты имеют концентрации, близкие к эвтектическим смесям, т. ё. концентрации, имеющие низкие температуры кристаллизации. Например, 75%-ная, 93,3%-ная серная кислота, олеум (SO3своб = 18,07%) имеют температуры кристаллизации, равные соответственно—41; —37,85; —17,05 G.
С увеличением концентрацииSO3 от 64,35% до 100% при кристаллизации образуются твердые растворы. Все сорта выпускаемой кислоты имеют концентрации, близкие к эвтектическим смесям, т. ё. концентрации, имеющие низкие температуры кристаллизации. Например, 75%-ная, 93,3%-ная серная кислота, олеум (SO3своб = 18,07%) имеют температуры кристаллизации, равные соответственно—41; —37,85; —17,05 G.
Серная кислота находит широкое применение в промышленности. Примерно половина производимой серной кислоты расходуется на производство удобрений и кислот. Она применяется для травления стальных изделий перед лужением, хромированием и т.п. очистки нефтепродуктов от непредельных и сернистых соединений, для производства ряда красителей, лаков и красок, лекарственных веществ, некоторых пластмасс, спиртов, ядохимикатов, синтетических моющих средств, искусственного шелка, в текстильной промышленности для обработки тканей или волокна перед крашением, а также дли производства крахмала, патоки и т. д.
Концентрированная серная кислота и олеум используют как водоотнимающее средства при производстве взрывчатых веществ (нитроглицерина, пироксила, тротила и др.). концентрировании азотной кислоты и т. д.
В связи с дальнейшим развитием промышленности и совершенство техники возникают новые отрасли потребления серной кислоты, поэтому ее производство с каждым годом растет (табл. 1).
Таблица 1 Производство серной кислоты в СНГ (в млн. т)
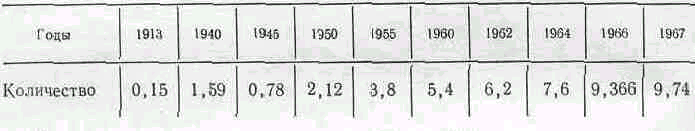
В промышленности серную кислоту получают нитрозным иконтактным способами. Независимо от способа производства сначала получают сернистый ангидрид SO2, который затем перерабатывают в серную кислоту.
4. Производство полимеров
Высокомолекулярные соединения получают из мономеров полимеризацией, сополимеризацией, поликонденсацией и методами привитой полимеризации и блокполимеризации.
Полимеризация — процесс образования высокомолекулярных соединений в результате взаимодействия мономеров с двойными связями в молекуле между собой или взаимодействия гетероциклов с размыканием колец

При проведении полимеризации совмещают воздействие тепла и химических веществ (катализаторы или инициаторы). Процесс полимеризации может вызываться облучением мономера γ-лучами, лучами рентгена, токами высокой частоты и фотохимически.
На процесс полимеризации большое влияние оказывает температура, которая резко повышает или скорость роста цепи, или обрыв цепи полимера, что ведет к уменьшению молекулярного веса полимера и средней степени полимеризации, поэтому поддерживают оптимальную температуру процесса.
В описанных процессах полимеризации, как правило, образуются полимеры аморфной структуры с неупорядоченным пространственным расположением боковых групп вдоль оси макромолекулы.
Применение комплексных катализаторов, состоящих из металлоорганических соединений А1(С2Н5)3 и хлоридов металлов переменной валентности (TiCI2, TiCl4), обеспечивает получение полимеров со строго линейной структурой и симметричной пространственной ориентацией. Такие полимеры получили название стереорегулярных. Они имеют большую прочность, плотность, высокую температуру плавления и легко ориентируются при вытягивании.
В промышленности применяют блочную, эмульсионную, лаковую, капельную или бисерную полимеризации.
При блочном методе мономер, очищенный от примесей и смешанный с катализатором или инициатором, подается в форму (сосуд), где нагревается. Для получения полимера с высокими свойствами необходимо строго поддерживать температуру. Полимер, получаемый в виде блока листа и т. п., из-за перегрева реакционной массы неоднороден.
При эмульсионной полимеризации мономер смешивается с инициатором и эмульгатором и с помощью мешалок превращается в мельчайшие капельки, взвешенные в другой жидкости — обычно в воде. Полученные эмульсии нагреваются до температуры начала реакции, и процесс полимеризации мономера в каждой мельчайшей капельке проходит самостоятельно. При этом можно легко отводить тепло, выделяемое в процессе полимеризации, поэтому получаемый полимер более однороден. Но эмульгатор трудно отделить от полимера, что затрудняет получение бесцветных материалов.
Лаковая полимеризация осуществляется в растворителе, смешивающемся с мономером и растворяющем образующийся полимер. Из полученного раствора полимер выделяют путем испарения растворителя или осаждением, или раствор может использоваться и качества лака. Кроме того, полимеризацию можно проводить в растворителе, в котором растворяется мономер, но не растворяется полимер. Образующийся полимер выпадает в твердом виде и отделяется фильтрованием. При этом получаются полимеры однородного состава, так как удается поддерживать определенную температуру процесса.
При капельной (суспензионной) полимеризации используются инициаторы, растворимые в мономере, но не растворимые в воде. Полимеризация проходит самостоятельно в каждой крупной капле мономера размером от 0,05 до 0,3 см в отличие от размера капли (от 103 до 104 см) при эмульсионной полимеризации. Образовавшийся полимер в виде твердых частичек, не растворимых в воде, осаждается.
Если при получении полимеров участвуют два различных ненасыщенных мономера, то такой процесс называется сополимеризацией. Метод сополимеризации позволяет увеличить число высокомолекулярных соединений, широко варьировать свойства получаемых продуктов. Процессы сополимеризации аналогичны процессам полимеризации.
Образование полимеров из мономеров при проведении процессов полимеризации или сополимеризации происходит без выделения побочных продуктов.
Поликонденсация — процесс образования высокомолекулярных соединений — полимеров путем реакции поликонденсации, с образованием полимеров и выделением побочных продуктов (H2O, эфиры, NH3 СО2 и др.). В реакцию поликонденсации вступают как одноименные мономеры, содержащие две различные реакционные группы, например аминокислоты (процесс гомополиконденсации), так и мономеры различного химического состава (процесс гетерополиконденсации).
При поликонденсации образующиеся полимеры могут иметь как линейное (полиамиды, полиэфиры, поликарбонаты), так и трехмерное строение (аминопласты, фенопласты). Скорость процесса поликонденсации и молекулярный вес полимера зависят от скорости вывода образующегося в процессе реакции побочного продукта, от температуры, концентрации реагирующих компонентов. Поликонденсацию проводят как с использованием катализаторов (аминопласты, фенопласты), так и без них (полиамиды). Процесс поликонденсации можно проводить в расплаве, по лаковому способу и на поверхности двух фаз.
Поликонденсацию в расплаве осуществляют при температуре 200—280°С в реакторе в атмосфере инертного газа. В конце процесса для полного удаления низкомолекулярных соединений в реакторе создается высокий вакуум. Этим способом получают полимеры в отсутствие растворителя.
Поликонденсация в растворе (мономеры растворяются в растворителе) проходит при малых скоростях, так как могут образовываться циклические соединения, и тогда затрудняется удаление низкомолекулярных продуктов реакции.
Полимеризация на поверхности раздела фаз проводится в несмешивающихся жидкостях, при этом взаимодействие мономеров между собой происходит быстро при низких температурах, так как выделяемые продукты выводятся из сферы реакции. Образующиеся высокоплавкие полимеры имеют высокий молекулярный вес. Такой способ получения полимера можно совместить с переработкой полимера в изделие.
Кроме основных методов получения высокомолекулярных соединений, находят применение методы блок- и привитой полимеризации.
В технике высокомолекулярные соединения являются основой для получения синтетических полимерных материалов. Большое значение из полимерных материалов имеют пластические массы, каучук и резина, химическое волокно, пленочные материалы, лаки, целлюлоза и др.
5.Производство химических волокон
Волокнами называют тела, длина которых во много раз превышает очень малые (микроны) размеры их поперечного сечения.
По происхождению волокна делят на природные натуральные и химические.
Химические волокна разделяют на искусственные, получаемые из природных полимерных соединений, и синтетические, получаемые из полимеров. Особую группу составляет стеклянное волокно.
Искусственные волокна делят на целлюлозные (вискозные, медно-аммиачные, ацетатные) и белковые (казеиновые, соевобовые), а синтетические — на карбоцепные и гетероцепные. К карбоцепным волокнам относят: хлорин, нитрон, политен, виньон, саран, винол и др., а к гетероцепным — полиамидные, полиэфирные, полиуретановые и др.
Для получения химических волокон применяются различные методы, имеющие много общего; но вместе с тем каждый метод имеет и свои особенности. Независимо от применяемого сырья технология изготовления волокон складывается из следующих стадий:
а) получение исходного материала;
б) приготовление прядильной массы;
в) формирование волокна;
г) отделка.
Высокомолекулярные соединения, применяемые для получения волокон, должны иметь высокую степень чистоты, растворяться или
плавиться.
Получение исходных материалов для изготовления синтетических волокон состоит из синтеза полимера — смолы, а для получения искусственного волокна необходимо отделение примесей от природных полимеров.
Приготовление прядильной массы для формирования волокон состоит из растворения полимеров в растворителях (спирте, Щелочи, ацетоне и др.) или расплавления смолы. Приготовленный раствор или расплав перед поступлением на формование очищают фильтрованием от примесей (примеси снижают прочность) и освобождают от пузырьков воздуха. В случае необходимости в раствор или в расплав вводят красители ддя придания волокну окраски, матовости и т. д
Формование волокна осуществляют по мокрому и сухому способам прядения из раствора и по сухому способу — из расплава. Независимо от способа формования приготовленную прядильную массу продавливают через фильеру (нитеобразователь), имеющую до 25000 отверстий диаметром от0,04ли< и выше. Образовавшиеся тонкие струйки раствора или нити расплава охлаждают или химически обрабатывают.
К искусственным волокнам относятся вискозные, ацетатные, медно-.аммиачные и др. Вискозное волокно находит наибольшее применение в технике. Для получения вискозного волокна прядильный раствор готовят из листов целлюлозы, обрабатываемой раствором едкого натра (18—20%), в результате чего образуется щелочная целлюлоза
![]()
6. Производство пластмасс
Пластические массы делят на простые (ненаполненные) и сложные (композиционные). Основу пластических масс составляет высокомолекулярное соединение — смола, которая при нагревании и давлении переходит в пластическое состояние, формуется под воздействием внешних сил и после охлаждения сохраняет полученную форму.
Простые пластмассы получают только из одной смолы, например полиэтилена.
Сложные пластмассы состоят из смолы, наполнителей, пластификаторов, красителей, стабилизаторов, отвердителей и др. Смола, являясь связующим веществом, придает смеси пластичность и формуемость.
Наполнители снижают стоимость пластмассы, придают или усиливают определенные механические и диэлектрические свойства, снижают горючесть изделий, улучшают внешний вид и т. п. В качестве наполнителей применяют порошковые (древесная, кварцевая мука, графит, тальк, асбест и др.) и волокнистые (ткани, асбестовое волокно и др.) материалы.
Пластификаторы повышают пластичность, эластичность композиции, но при увеличении их количества прочность на разрыв и сжатие резко снижается. В качестве пластификаторов используют малолетучие вещества (камфору, касторовое масло, дибутклфталаты. трикре-зилфосфат). Красители придают пластическим массам желаемую окраску. Они должны хорошо смешиваться и совмещаться со смолой и выдерживать воздействие температуры, воды и т. п., сохраняя цвет как в процессе формования, так и при применении изделия.
Отвердители — вещества, способные превращать линейную структуру полимера в результате сшивания макромолекул в трехмерную структуру. К ним относятся уротропин, гексаметилентетрамин и др.
Кроме перечисленных веществ, в состав сложной пластмассы вводят стабилизаторы, способствующие сохранению первоначальных свойств; смазки, облегчающие прессование; порошкообразователи — для получения пено- и поропластов.
Пластические массы (пластмассы) имеют низкую плотность (900— 1750 кг/м3), высокие механические и хорошие электроизоляционные свойства, высокую водостойкость, теплостойкость и химическую стойкость, красивый внешний вид и т. д. Стоимость некоторых пластических масс не превышает стоимости цветных, а в некоторых случаях и черных металлов при расчете на 1 м3 материала. Такие технико-экономические преимущества пластических масс перед другими материалами обусловили широкое их применение. Но пластмассы имеют низкую теплостойкость (60—300° С), подвержены “старению”, что снижает их свойства.
Промышленность выпускает большое количество синтетических смол и пластмасс на их основе, поэтому для примера рассмотрим технологию получения и свойства наиболее важнейших смол и пластмасс на их основе.
Полимеризационные смолы для получения пластмасс используют без наполнителей. Они термопластичны, обладают хорошими диэлектрическими свойствами, высокой ударной прочностью (кроме полистирола), химически стойки, но большинство из них имеет низкую теплостойкость.
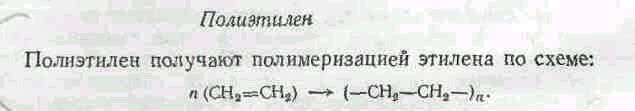
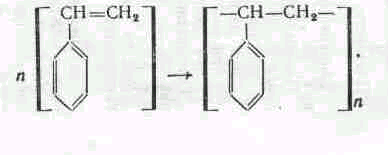
Сырьем для получения полистирола является стирол (С6Н5СН=СН2). Полимеризацию стирола проводят лаковым, эмульсионным и блочным способами. Процесс полимеризации проходит по следующей схеме:
7.Синтезы на основе ацетилена
Ацетилен СН=СН — газ, легко вступающий в самые различные химические реакции с образованием многочисленных соединений, используемых при получении волокон, каучуков, смол и т. д. Например, из ацетилена получают ацетальдегид, этиловый спирт, бутадиен, этил-ацетат, хлористый винил, винилацетат, хлоропрен, акрилонитрил и др. Он используется для получения высоких температур (3200° С) при сварке и резке металлов.
Ацетилен получают путем обработки карбида кальция водой (СаС2 + 2Н2О ->Са(ОН)2 + С2Н2) или путем термоокислительного крекинга при 1400—1500° С различных углеводородов (СН4, С2Н4 и до.)
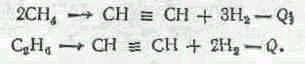
Для примера рассмотрим синтез ацетальдегидана основе ацетилена.
Ацетальдегид СНзСНО — альдегид уксусной кислоты, летучая жидкость с резким запахом, хорошо смешивается с водой и спиртом. Он используется для получения уксусной кислоты.
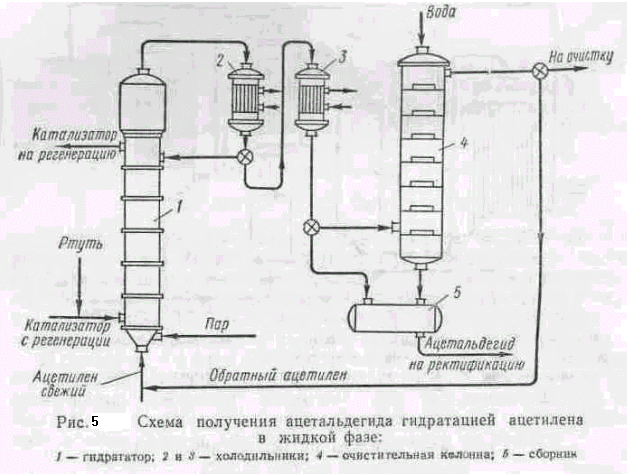 В промышленности ацетальдегид по одной из схем (рис.5) получают следующим образом. Очищенный от примесей ацетилен, смешанный с циркуляционным газом, непрерывно подается в гидрататор 1, в котором находится нагретая до 80—100° С жидкость, содержащая сульфаты железа и ртути (в 1 л Н2О 200 е H2S04, 0,4 г Hg, 40 г окислов железа). Ацетилен, барботируя через жидкость, переходит в ацетальдегид по реакции:
В промышленности ацетальдегид по одной из схем (рис.5) получают следующим образом. Очищенный от примесей ацетилен, смешанный с циркуляционным газом, непрерывно подается в гидрататор 1, в котором находится нагретая до 80—100° С жидкость, содержащая сульфаты железа и ртути (в 1 л Н2О 200 е H2S04, 0,4 г Hg, 40 г окислов железа). Ацетилен, барботируя через жидкость, переходит в ацетальдегид по реакции:
СН = СН + Н2О --> СНзСНО + 1416 кдж.
Степень перехода ацетилена в ацетальдегид составляет 50—60%. Газы, содержащие ацетальдегид, ацетилен и примеси, поступают на охлаждение сначала в холодильник2, где частично конденсируются пары воды, и конденсат возвращается в гидрататор 1, а затем в холодильник 3, где конденсируются пары ацетальдегида и воды, собираемые в сборнике 5 и направляемые на ректификацию (на схеме не показано).
Газы, содержащие ацетилен, поступают в колонну 4, орошаемую водой, где из них извлекаются остатки ацетальдегида, и снова возвращаются в процесс. Для очистки оборотного газа от окислов углерода и азота часть его (10%) непрерывно отбирается из цикла и направляется на очистку. Выход ацетальдегида составляет примерно 96% от теоретического. Так как пары ртути и ее соединения ядовиты, начинают применяться не ртутные катализаторы в виде окислов Zn, Mg,Ni, Со, Cr.
Уксусная кислота находит применение в текстильной, фармацевтической, парфюмерной промышленности, для получения сложных эфиров, в производстве ацетилцеллюлозы, уксусного ангидрида, ацетона, ацетатного шелка, каучука, пластических масс и т. д.
Раньше кислоту получали сухой перегонкой древесины, брожением этанола, а в настоящее время — окислением ацетальдегида или гидратацией кетона.
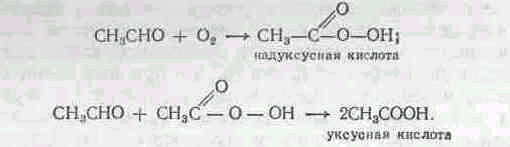
Окисление ацетальдегида кислородом воздуха происходит в присутствии солей марганца при темпеатуре 60—70° С. Так как надуксусная кислота может взрываться, то парогазовая смесь разбавляется азотом. Образование СНзСООН идет по следующей схеме:
Для очистки от примесей синтетическую кислоту подвергают перегонке. Техническая кислота после перегонки содержит 97—99% СНзСООН, 0,1—0,5% НСООН, 0,5—2% Hp.
Безводная уксусная кислота плотностью 1049 кг/м3 (20° С) застывает при 16,6° С, кипит при 118,1° С. Она хорошо смешивается с во" дои и многими органическими жидкостями. При попадании на кожу вызывает ожоги.
8. Подготовка угля к коксованию
Коксование—процесс сухой перегонки каменных углей при их нагревании до 900—1050° С без доступа воздуха. В результате сложных физических и химических превращений образуется твердый, спекшийся продукт — кокс и прямой коксовый газ.
Кокс используют в металлургии, литейном производстве, для получения электродов, карбида кальция и т. д.
Прямой коксовый газ содержит каменноугольную смолу, сырой бензол и другие продукты, поэтому его перерабатывают с получением ценных химических веществ.
Сырьем для получения кокса служат спекающиеся (коксующиеся) угли марки К, которых в недрах земли содержится мало Для расширения сырьевой базы для коксования применяют смесь — шихту, состоящую из коксующихся углей и углей других марок, мало содержащих серы и фосфора, которые при коксовании остаются в коксе и снижают его качество.
Перед поступлениемна коксование угли тонко измельчаются— зерен размером менее 3 мм должно быть 85—90%.
Процесс коксования осуществляется в коксовой печи, представляющей собой камеру, выложенную огнеупорным (динасовым) кирпичом. Камеры по 60—70 шт. соединяются между собой в коксовые батареи; между ними имеются пространства (простенки), в которых сжигается генераторный или коксовый газ. Температура в простенках печи достигает 1400° С. Так как огнеупорный кирпич и уголь являются плохими проводниками тепла, а для получения кокса требуется нагреть шихту до 900—1050° С, камеры делают в виде узких каналов — шириной — 0,4 м, длиной — 13—14 м, высотой — 4, 4,5 м. В камеру загружают до 15 т угля.
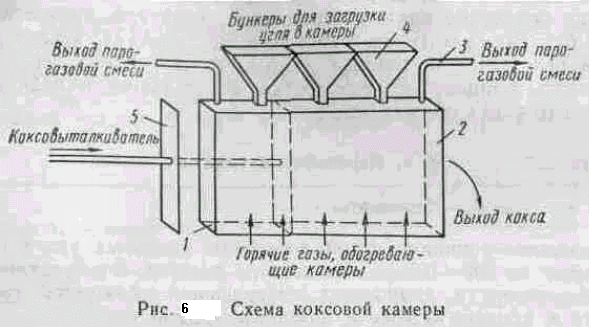 На рис. 6 показана схема коксовой камеры. Она имеет две торцовые стороны — коксовую, куда выталкивается из камеры кокс (коксовый пирог), и машинную — для ввода в камеру коксо-выталкивателя 5, представляющего собой пластину с размерами, немного меньшими, чем у сечения камеры. При коксовании машинная и коксовая стороны камеры плотно закрываются дверцами / и 2. В своде камеры имеются отверстия, через которые загружается шихта с помощью загрузочного вагона с бункерами 4, течки которых устанавливаются над отверстиями в своде камеры. Вагон перемещается по рельсовому пути, расположенному над коксовыми камерами, и обслуживает десятки камер. После загрузки шихта в камере разравнивается, загрузочные отверстия закрываются и начинается процесс коксования.
На рис. 6 показана схема коксовой камеры. Она имеет две торцовые стороны — коксовую, куда выталкивается из камеры кокс (коксовый пирог), и машинную — для ввода в камеру коксо-выталкивателя 5, представляющего собой пластину с размерами, немного меньшими, чем у сечения камеры. При коксовании машинная и коксовая стороны камеры плотно закрываются дверцами / и 2. В своде камеры имеются отверстия, через которые загружается шихта с помощью загрузочного вагона с бункерами 4, течки которых устанавливаются над отверстиями в своде камеры. Вагон перемещается по рельсовому пути, расположенному над коксовыми камерами, и обслуживает десятки камер. После загрузки шихта в камере разравнивается, загрузочные отверстия закрываются и начинается процесс коксования.
Для отвода паро-газовой смеси из камеры стояк 3 соединяется с газопроводом.
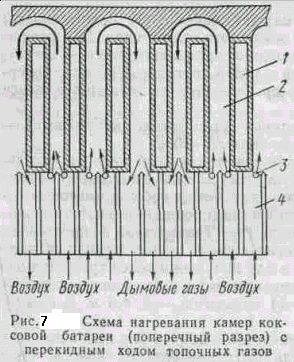 На рис. 7 показана схема нагревания шихты в камерах коксовой батареи (поперечный разрез) с перекидным над сводами камер ходом топочных газов. Воздух, поступающий на горение горючих газов, предварительно нагревается в регенераторах 4 и смешивается с газом, поступающим из отверстий 3 в простенках 2, расположенных между камерами /. В простенке 2 происходит сгорание газообразного топлива, и горячие дымовые газы огибают камеру, подогревают ее с другой стороны и уходят через регенераторы тепла в дымовую трубу.
На рис. 7 показана схема нагревания шихты в камерах коксовой батареи (поперечный разрез) с перекидным над сводами камер ходом топочных газов. Воздух, поступающий на горение горючих газов, предварительно нагревается в регенераторах 4 и смешивается с газом, поступающим из отверстий 3 в простенках 2, расположенных между камерами /. В простенке 2 происходит сгорание газообразного топлива, и горячие дымовые газы огибают камеру, подогревают ее с другой стороны и уходят через регенераторы тепла в дымовую трубу.
Через каждые 20—30 мин поток газа и воздуха переключают на нагретые топочными газами регенераторы и поток газов обогревает обратную сторону камеры.Это обеспечивает равномерный нагревкамеры с обеих сторон. На заводах применяют различные системы обогрева камер; пребывание шихты в камере 13—17 ч. Выделяющийся при коксовании в камерах прямой коксовый газ отсасывается воздуходувкой и подается на переработку.
По окончании процесса коксования разгрузка камерпроводитсяпоочередно. После разгрузки камеры торцовые стороныее закрываются и цикл работы повторяется.
Из 1 т шихты с влажностью 6% в процессе коксования получают в среднем следующие продукты, кг:

Переработка прямого коксового газа
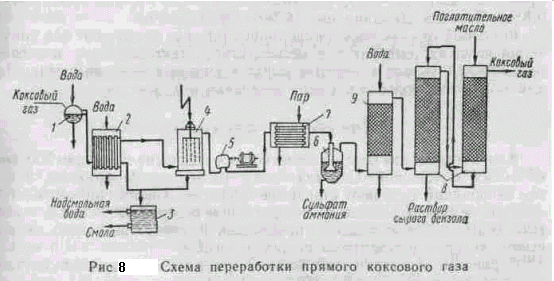 Парогазовую смесь, выходящую из коксовой камеры, называют прямым коксовым газом. В 1 м3 газа, кроме Н2, СН4, СО и газообразных углеводородов, содержится: смолы 80—130 г, бензольных углеводородов 30—40 г, аммиака 8—13 г, сероводорода и других сернистых соединений 6—25 г, цианистых соединений — 0,5—1,5 г, паров воды 250—450 г, твердых частиц 15—35 г. Такой газ подвергают переработке по схеме, приведенной на рис. 8.
Парогазовую смесь, выходящую из коксовой камеры, называют прямым коксовым газом. В 1 м3 газа, кроме Н2, СН4, СО и газообразных углеводородов, содержится: смолы 80—130 г, бензольных углеводородов 30—40 г, аммиака 8—13 г, сероводорода и других сернистых соединений 6—25 г, цианистых соединений — 0,5—1,5 г, паров воды 250—450 г, твердых частиц 15—35 г. Такой газ подвергают переработке по схеме, приведенной на рис. 8.
Прямой коксовый газ, выходящий из камеры при температуре 700—800° С, поступает в газосборник /, где охлаждается до 80° С водой; при этом из газа частично конденсируется смола и твердые вещества. Для дополнительного выделения смолы газ охлаждают в холодильнике 2 до 20—30° С. Сконденсировавшаяся смола и надсмольная вода из газосборника 7 и холодильника 2 поступают в сборник 3, где разделяются на три слоя: нижний — твердые вещества, средний — смола” верхний — надсмольная вода. В надсмольной воде содержится аммиак. Для окончательного выделения из газа туманообразной смолы газ из холодильника 2 поступает в электрофильтр 4, где из него выделяется смола, стекающая в сборник 3. Для продвижения прямого коксового газа через систему аппаратов очистки применяется турбогазодувка 5. Пройдя турбогазодувку, газ нагревается в подогревателе 7 до 60— 70° С и поступает в сатуратор 6 — аппарат барботажного типа, в котором находится 76—78% H^SO^. Аммиак, содержащийся в газе, реагирует с HoS04 с образованием сульфата аммония;
![]()
Образовавшийся сульфат аммония выпадает в осадок, отделяется от раствора, сушится и используется в качестве удобрения. Затем газ охлаждается до 20—25° С в холодильнике 9 и поступает в башни с насадкой 8, орошаемые каменноугольным маслом (фракция при перегонке смолы, кипящая при 230—300° С), которое извлекает из газа бензол, толуол, ксилол и др.
Раствор сырого бензола подвергается перегонке, в результате чего отгоняется бензол и его гомологи, а масло после охлаждения снова возвращается на орошение башен 8. Освобожденный от примесей коксовый газ называется обратным. Он очищается от соединений серы. Обратный коксовый газ в основном состоит из водорода (54—59%), метана (23—28%), окиси углерода (5—7%), углеводородов (2—3%) и примесей: азота (3—5%), углекислоты (1,5—2,5%), кислорода (0,3— 0,8%). Теплота сгорания его 16750—17200 кдж/м3.
Коксовый газ как высококалорийное топливо применяют для получения высоких температур в металлургии, стекловарении, коксовании; его используют в качестве сырья в химической промышленности для получения водорода, сажи, ацетилена и т. д.
9. Получение синтезированного газа
Химические методы переработки нефти проводят при высоких температурах без катализатора (термический крекинг), при высоких температурах в присутствии катализатора (каталитический крекинг), в присутствии водорода, при высокой температуре и давлении (гидрокрекинг) и др. Благодаря высокой температуре происходит расщепление молекул углеводородов. Кроме того, в результате вторичных процессов образуются молекулы новых соединений, которые не содержатся в нефти или в нефтепродуктах.
Рассмотрим процесс расщепления составляющих нефти при нагревании на примерах.
При нагревании нефти сначала расщепляются углеводороды парафинового ряда с длинной цепью

По мере повышения температуры разрыв цепи углеводородов сдвигается к краю цепи, вплоть до метана СН4, т. е. С14Н30 а С13Н26 + СН4, а при температуре выше 820° С метан разлагается на углерод и водород СН4 --> С + 2Н2
Нафтеновые углеводороды при нагревании дегидрируются, образуя ароматические углеводороды:
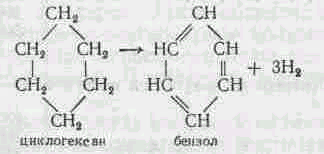
Ароматические углеводороды более устойчивы к нагреванию, поэтому они почти не изменяются. Непредельные углеводороды, образующиеся в процессе распада, в значительной степени вступают в реакцию полимеризации или циклизации, образуя ароматические и другие сложные соединения. Чем выше температура крекинга, тем выше скорость реакции и больше образуется газообразных продуктов. Применение давления 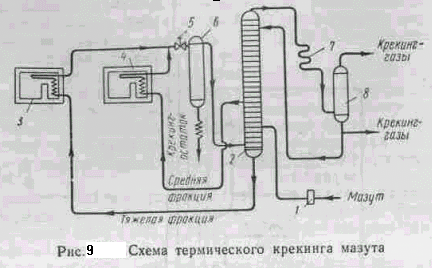 затрудняет процесс расщепления и благоприятно влияет на вторичные реакции. Крекинг ведут для получения бензина и газов.
затрудняет процесс расщепления и благоприятно влияет на вторичные реакции. Крекинг ведут для получения бензина и газов.
Термический крекинг в смешанной фазе (жидкой и паровой) проводят под давлением до 70 am при температуре 350—500° С. На рис.9 показана схема крекинга мазута. Мазут насосом 1 подается на одну из нижних тарелок ректификационной колонны 2, где смешивается с тяжелой фракцией. Затем смесь подается в трубчатую печь 3, где нагревается до температуры 470—480° С. Из средней части колонны 2 выводится более легкокипящая фракция, которая нагревается в трубчатой печи 4 до 500—510°. Давление в печах поддерживается 50—70 am (5—7 Мн/м2). Продукты крекинга из печей 3 и 4 проходят редукционный вентиль 5 и поступают в испаритель 6, где происходит отделение паров от крекинг остатка, который выводится из испарителя. Пары из испарителя направляются в ректификационную колонну на разделение. Пары бензина и газы проходят конденсатор 7 и сепаратор 8, где они разделяются.
Выход продуктов при крекинге следующий: крекинг-бензин 30— 35%, крекинг газы 10—15%, крекинг-остаток 50—56%. Крекинг газы содержат этилен, пропан, пропилен, бутан, бутилен и др. Они служат ценным сырьем для синтеза органических соединений. Крекинг-остаток служит котельным топливом.
Парофазный крекинг — пиролиз проводят при температуре 670— 720° С и атмосферном давлении. В процессе пиролиза жидкие продукты обогащаются ароматическими соединениями, а газы — непредельными углеводородами. Пиролиз проводится с целью получения сырья для химической промышленности.
Каталитический крекинг проводится в паровой фазе при 450-500е С и давлении 0,5-1,0 am (0,05-0,1 Мн/м2) в присутствии алюмо-силикатных катализаторов, представляющих собой твердые высоко. пористые вещества. Катализаторы адсорбируют углеводороды и на поверхности происходят реакции расщепления. Одновременно мот проходить реакции ароматизации. При каталитическом крекинге наряду с жидкими продуктами (вы. ход бензина — 70%) образуются газы (12—15%) и кокс (4—6%) Кокс откладывается на поверхности катализатора и снижает его активность. Для выжигания кокса через катализатор при температуре 550— 600 С пропускают воздух.
В печи 1 подаваемое на крекинг сырье нагревают до 350—360° С и затем направляют в реактор 2, в который из бункеpa 3 поступает зернистый катализатор. Под действием собственного веса катализатор опускается в низ реактора и перед поступлением в регенератор (самотеком) обрабатывается паром. Продукты крекинга из реактора 2 направляются на разделение в ректификационную колонну (на схеме не показана). Для регенерации катализатора сверху в регенератор 4 воздуходувкой 5 подается воздух, который реагирует с коксом, расположенным на поверхности катализатора, очищая его. Образовавшиеся дымовые газы выводятся снизу генератора. Из нижней части регенератора 4 катализатор захватывается сжатым воздухом, подаваемым воздуходувкой 7, и по трубе 6 направляется в бункер 3 и снова возвращается в реактор 2.
Для уменьшения отложений кокса на катализаторе применяют крекинг под давлением в присутствии водорода. Такой процесс получил название риформинга. Наибольшее применение риформинг находит с платиновыми, хромовыми и молибденовыми катализаторами. Для риформинга используют фракции легких нефтепродуктов. Процесс проводится под давлением 40—70 am (4—7 Мн/м2) при температуре 480—520° С.
В процессе риформинга происходит образование ароматических углеводородов, которые улучшают качество бензинов. Газы риформинга содержат СН4, C2H6, С3Н8, С4Н10. Их используют для синтеза органических соединений.
Список использованной литературы
- Баринов Н.А. Технология металлов. Металлургиздат.1963
- Сидоров И.А. Основы технологии важнейших отраслей промышленности, Москва, “высшая школа”, 1971
- Кован В.М. (и др.) Основы технологии машиностроения “Машиностроение”, 1965
- Никифоров В.М. (и др.) Технология важнейших отраслей промышленности, ч.1, изд. ВПШ при ЦК КПСС, 1959
- Данилевский В.В. Технология машиностроения. “Высшая школа”, 1965